Scientific confidence: Very High

You are suspended inside liquid water — not observing it, but enclosed within it, surrounded in every direction by pale-blue translucent oxygen spheres pressing close at oxygen-to-oxygen spacings of just 2.75 Å, so near that the concept of empty space becomes meaningless. Each bent molecule carries two bright proton lobes angled at 104.5°, and between neighboring pairs, cyan hydrogen bonds — flickering threads of shared electron density — pulse and snap on picosecond timescales, too fast for any single event to register as discrete motion, perceived instead as a collective, living tremor running through the entire lattice. This is liquid water at 300 K: not the featureless solvent of intuition, but a dense, constantly rewiring hydrogen-bond network in which every molecule simultaneously donates and accepts bonds, achieving no fixed crystal order yet never collapsing into chaos. Beyond three or four molecular diameters, successive shells of molecules thicken into a deep cerulean haze, compressing the visual field into an intimate radius of resolution — only a handful of fully-formed neighbors visible before the world dissolves into indigo depth. The light has no source; it diffuses outward from the electron clouds of oxygen itself, scattering through overlapping shells until the entire environment glows cold and aqueous, simultaneously claustrophobically close and boundlessly infinite, the most abundant molecule on Earth revealed as a ceaselessly breathing, alien cosmos.

The world underfoot is an infinite plain of warm hexagonal discs, each one tilted at a sharp alternating angle to its neighbor in a herringbone weave that repeats with hypnotic exactness in every direction, like a vast tiled floor laid by some crystallographic intelligence working at a scale where cobblestones are made of electron density rather than stone. These are benzene molecules frozen into their monoclinic lattice at 175 K, each flat aromatic ring held in place not by covalent bonds to its neighbors but by the subtle geometry of edge-to-face CH–π interactions — one molecule's hydrogen edge leaning toward another's π face across a gap of roughly 3.5 ångströms, a distance so intimate that the ghostly violet-amber halos of delocalized π-electron cloud hovering above and below each molecular plane nearly overlap, pooling faintly at the closest approach like shared breath. The cold is not warmth but absolute stillness: bond vibrations reduced to their irreducible quantum minimum, every disc locked in crystallographic exactitude, the entire landscape held in a frozen cathedral geometry that recedes through softly glowing amber-indigo corridors toward a luminous infinity. Here and there a dark socket interrupts the perfection — a vacancy defect where a single molecule is absent, its neighbors leaning fractionally inward, their π halos fraying at the edges as though registering the disruption in the only language available to them: a slight asymmetry in the otherwise flawless periodic order.

At the absolute midplane of a fluid DPPC bilayer — a membrane just four nanometers thick from headgroup to headgroup — the immediate world is a dim, waxy charcoal expanse where the methyl termini of opposing fatty acid chains barely graze one another across the hydrophobic void, a boundary so molecularly sparse it registers as near-silence after the dense architecture above. Rising outward through either leaflet, long sinuous CH₂ corridors streak upward like brushed pewter pillars, occasionally interrupted by the warm yellow-green elbow of an unsaturated double bond that kinks the chain geometry and scatters ambient light differently than its saturated neighbors — each such bend a structural defect that fluidizes the bilayer and keeps it from crystallizing at body temperature. The glycerol-ester stratum announces itself as an amber-translucent geological seam, electron-dense ester linkages glinting like resin nodules at the precise frontier where hydrophobic territory surrenders to polar chemistry. Beyond that boundary the world erupts into the crowded, electrostatically charged headgroup region: phosphate spheres burn deep saturated orange while choline nitrogens glow cool sky-blue above them, both leaflets incessantly jostled by the cold, glittering hydrogen-bond network of bulk water whose picosecond-scale restructuring reads as a perpetually forming and dissolving crystalline frost pressing in from either aqueous face.

You stand at the bottom of a canyon that has no geological age — carved not by water or wind but by the precise logic of base-pairing and phosphodiester bonds, its walls built fresh at every cell division and read continuously by proteins that must find their footing on this same uneven terrain. The groove enclosing you is the major groove of B-form DNA: 22 ångströms wide and lined on both sides by the sugar-phosphate backbone, whose deoxyribose rings arch upward in bronze pentagonal frames while the phosphate groups project outward as rust-orange tetrahedra, each one slicked with an ordered shell of water molecules arranged in pearl-like hydrogen-bonded chains — the spine of hydration that stabilizes the helix and helps transcription factors recognize their binding sites without ever touching the bases directly. Beneath your feet, the groove floor is a mosaic of stacked base-pair discs — teal adenine against sienna thymine, forest-green guanine against lavender cytosine — separated by 3.4-ångström π-stacking gaps, those hairline shadows where aromatic electron clouds overlap just enough to contribute several kilocalories per mole of stabilizing energy to the entire double-helical architecture. The negatively charged phosphate backbone floods the atmosphere with a deep cobalt electrostatic field, a volumetric blue fog that grows denser toward the walls and draws counterions and water into the groove, making the medium around you not empty space but a dense, electrostatically sculpted aqueous environment where every surface trembles with thermal vibration at frequencies measured in femtoseconds — geometry that is real and precise but never, even for an instant, entirely still.

You are floating along the central axis of a protein α-helix, suspended in a dense amber solvent that blurs the most distant turns into warm rose-gold haze — each backbone turn spanning roughly 5.4 Ångströms in pitch, the full structure uncoiling through some thirty repeating units ahead of you before curving out of sight. The spiral wall around you is an architectural record of quantum mechanical precision: deep-crimson carbonyl oxygens and pale backbone nitrogens lock together in hydrogen bonds bridging every fourth residue at a distance of just 2.06 Å, the stored electrostatic energy of each bridge bleeding a faint magenta luminescence into the turbid medium, while the covalent bonds themselves radiate cool blue-white electron density wherever orbital overlap is densest. Projecting outward from this backbone scaffold, leucine and isoleucine side chains thrust toward you like crystalline yellow-green thorns, their branching tetrahedral carbon geometries repelling the aqueous medium with hydrophobic surfaces that leave a thin dry shimmer in the surrounding solvent — an arrangement not accidental but shaped by billions of years of evolutionary pressure toward the compactly folded, thermodynamically stable helix you now inhabit. The entire structure trembles imperceptibly at picosecond timescales, each atom mid-oscillation, every hydrogen bond forming and breaking in a rhythm too fast for any clock but the molecule's own, yet the architecture persists — a self-reinforcing spiral of geometry, charge, and thermal motion that is simultaneously alien in its submicroscopic exactness and overwhelmingly, viscerally present.

Looking straight down the crystallographic c-axis, the viewer is drawn into an infinite honeycomb cathedral of pale turquoise and deep blue-black, where each water molecule sits anchored at its lattice point like a frosted lantern, connected to four nearest neighbors by gently luminous hydrogen bonds spanning just 2.76 Å in perfect tetrahedral geometry. The commanding feature of this world is the series of hexagonal voids punched clean through the lattice at regular intervals — not empty in any casual sense, but structural absences born of tetrahedral bonding geometry that refuses to close-pack, and it is precisely these channels that give ice its anomalously low density compared to liquid water. At each bond, a subtle double shadow betrays proton disorder: the hydrogen is not fixed but statistically displaced toward one oxygen or the other, the probabilistic blur that Linus Pauling recognized in 1935 as an intrinsic residual entropy frozen into the crystal. The lattice-light seems to originate from within the structure itself — cold, sourceless, blueish-white at the nearest molecular nodes and deepening to saturated teal-indigo as crystallographic depth accumulates — while a faint softness at every molecular contour speaks to the thermal vibrations still alive at −10°C, causing the most distant visible layers to dissolve into a luminous, quietly trembling haze.

The viewer stands at the base of an infinite colonnade of coronene columns rising through an organic crystal, each aromatic disc roughly 9 ångströms across and separated from its neighbour by only 3.4 ångströms — the same interval that governs the interlayer spacing of graphite, a distance set by the equilibrium between π-electron repulsion and dispersive van der Waals attraction. Coronene is a polycyclic aromatic hydrocarbon of twelve fused benzene rings whose sp²-hybridized carbon framework forces every atom into a single plane, allowing the π-electron clouds above and below each disc to overlap with those of adjacent molecules in the stack, producing the shared luminous halos visible at every inter-disc gap — genuine electron density merging into a quasi-continuous conjugated pathway along which polarons, charge carriers dressed in local lattice distortion, hop between molecules in bursts that appear here as electric-orange flashes. The hexagonal crystal packing places each column within van der Waals contact of its neighbours, the inter-column space filled not with vacuum but with the diffuse, opalescent haze of weak dispersive interactions — forces negligible at human scales yet here the sole architecture holding an entire ordered solid together. Standing inside this structure is to inhabit a world where gravity has no jurisdiction, where thermal vibrations shake every disc by a fraction of a bond length sixty times per picosecond, and where the amber glow of delocalized aromatic electrons is not metaphor but a direct translation of quantum mechanical reality into light.

You are standing inside a living tunnel carved not from stone but from the coiled architecture of RNA itself — the exit channel of a ribosomal large subunit, a passage barely ten nanometers long and, at its narrowest constriction, scarcely wider than a single extended amino acid side chain. The walls press inward in deeply ribbed columns of double-helical rRNA, their phosphate-sugar backbones running in longitudinal ridges of cobalt and deep teal, punctuated at every helical turn by magnesium ions that catch the ambient chemical luminescence and throw back hard yellow-white glints, each one neutralizing a node of negative phosphate charge like a rivet holding pressurized hull plates together against the implicit crush of molecular electrostatics. Through the center of this channel snakes the nascent polypeptide chain, rendered in warm amber as it emerges residue by residue from the peptidyl transferase center high above — its backbone tracing sinuous kinks and early helical coils already consolidating their hydrogen bonds, gossamer threads of luminescence barely visible along its length as secondary structure nucleates before the chain has even left the tunnel. At the darkened periphery beyond the innermost RNA wall, teal pulses flicker and die in femtoseconds — GTP hydrolysis events translated here into brief aqueous luminescence, the thermodynamic currency of translation expressed as cold bioluminescent light dissolving instantly into the dense, invisible press of solvent that occupies every ångström of unclaimed space.
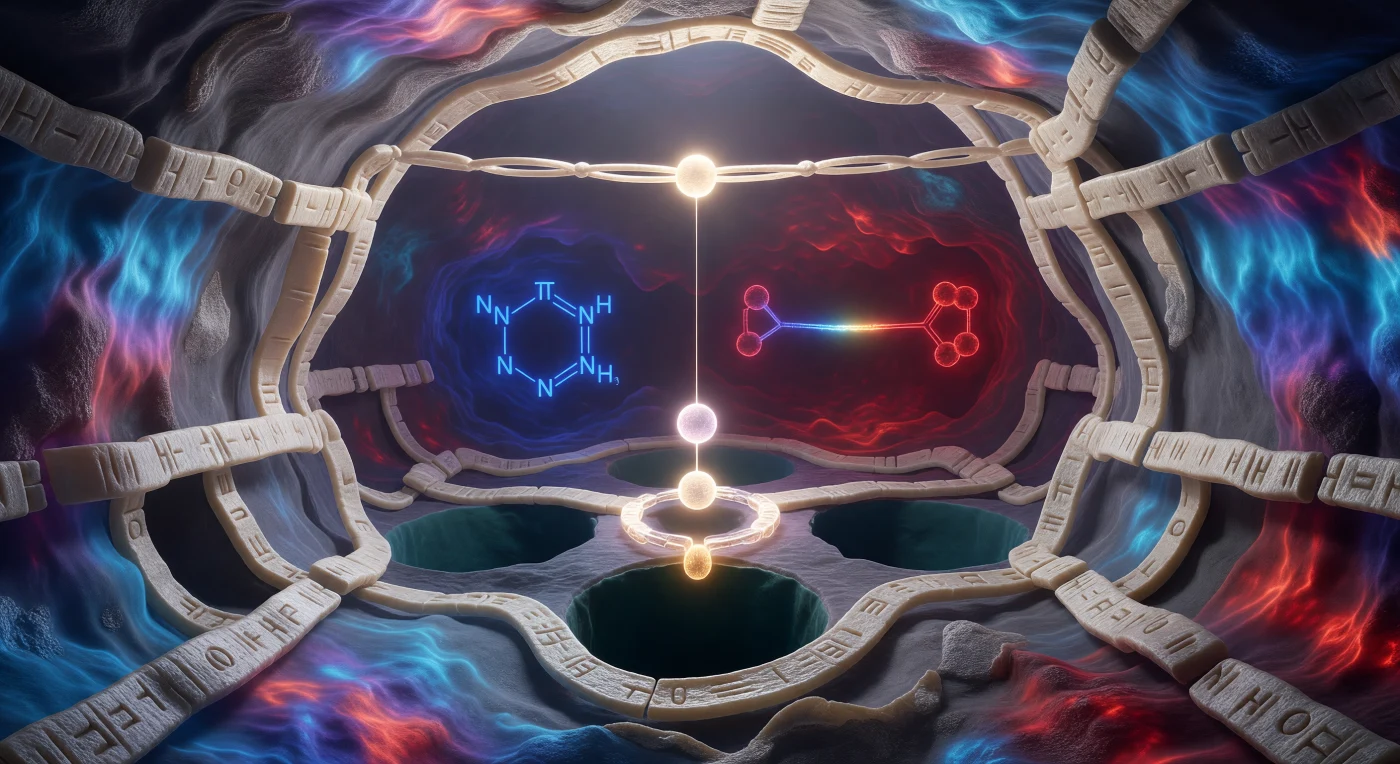
You are standing inside a catalytic pocket so small that the entire chamber spans barely fifteen ångströms from wall to wall, yet the architecture here has been refined by four hundred million years of evolutionary pressure into one of biochemistry's most precise reaction environments. The walls — pale ivory ribbons of beta-sheet and alpha-helix, their surfaces granular with the van der Waals radii of individual residues — glow in a continuous thermal electrostatic map: cool cerulean at the entrance bleeding through violet into deep scarlet at the rear, where the aspartate of the catalytic triad anchors its twin oxygens in burgundy charge against the back wall, stabilizing the entire network through electrostatic communication rather than covalent contact. Midway through the cavern, a histidine imidazole ring floats in electric cobalt blue, its π-electron cloud humming between two tautomeric states as a proton shuttle; directly below the substrate's scissile bond — which arches overhead like a luminous bridge, its carbonyl carbon suspended at approximately three ångströms of charged tension above the active-site floor — the hydroxyl oxygen of Serine 195 blazes white-gold, its lone pairs poised to launch a nucleophilic attack that will break the peptide bond in microseconds. Everything here vibrates: bond lengths oscillate on femtosecond timescales, side chains tremble through nanosecond fluctuations, and the ghost of recently expelled solvent water charges the pocket with an electric expectancy that is less metaphor than measurable electrostatic fact.

You stand at the absolute edge of a world one atom thick — the graphene sheet extending before you as a razor-dark discontinuity against vacuum, its boundary not quite a surface in any classical sense but a crystallized mathematical plane where carbon atoms line up like sentinels along a frontier that has no depth whatsoever, their dangling bonds glowing faintly amber where the hexagonal lattice terminates and electron clouds dissolve into emptiness. Wheeling upward to look down across the top-down expanse, the entire visible universe becomes a silver-blue hexagonal chainmail of sp²-hybridized carbon rings, each 1.42 Å carbon-carbon bond a node of elevated electron density within a continuous delocalized π-electron cloud that lends the whole infinite surface a cold metallic iridescence reminiscent of polished platinum — this is not a thin coating of metal but something stranger, a purely two-dimensional conductor where electrons flow through conjugated orbitals spread simultaneously above and below a plane of zero measurable thickness. Long-wavelength flexural phonons — quantized rippling vibrations with wavelengths stretching from tens to hundreds of nanometers — roll across the flatland in slow frozen swells, bowing the otherwise mathematically perfect lattice into gentle topographic undulations that distort the hexagonal symmetry at their crests like a geometric tide. Then, mid-distance, the perfection ruptures: a Stone-Wales defect, born from a single 90° bond rotation during formation, fuses one pentagon to one heptagon and traps localized electronic states that glow a distinct warm orange against the surrounding cold blue — a topological scar, an ember pressed into frost, radiating its energy signature outward through a dozen lattice rings before the crystalline order reasserts itself and the silver-blue chainmail resumes its silent, flawless extension toward every horizon.

You are hovering at a distance smaller than a single nanometer from a sodium cation, close enough that the ion's electrostatic field is the dominant force shaping everything around you — no gravity, no wind, only the relentless pressure of concentrated positive charge radiating outward and bending the surrounding water into rigid, symmetrical submission. Six water molecules lock into an octahedral cage precisely 2.36 ångströms from the ion's nucleus, their electronegative oxygen faces wrenched inward by the Na⁺ field, their dipoles so strongly aligned that the hydrogen-bond order here is measurably higher than in bulk water — a genuine phase-like transition imposed by a single charged particle. Beyond this inner sanctum, a softer second shell of twelve to eighteen water molecules partially yields to the same ordering influence at roughly 4.5 ångströms, their alignment already fraying at the edges where thermal energy — on the order of kT, some 25 meV at room temperature — begins to compete with electrostatic coherence. Past 7 ångströms, the bulk hydrogen-bond network resumes its chaotic, picosecond-timescale restructuring, each molecule breaking and reforming bonds with its neighbors in a dense, restless churn that carries no memory of the ion's authority — a reminder that at this scale, order is always local, always borrowed, and always temporary.

You are pressed into the amber heart of a folded protein, surrounded on every side by hydrocarbon chains and aromatic rings packed so tightly — at a density rivaling that of cut gemstone — that the nearest molecular surface is never more than a few ångströms away. Leucine and isoleucine branches interlock in warm ivory and beige, their methyl groups touching in soft van der Waals contacts at 3.5–4.0 Å, while phenylalanine rings slot between them like pale gold discs held in offset parallel planes by delocalized π-electron interactions. To one side, the bicyclic indole ring of a tryptophan residue radiates a deep teal-blue luminescence — its conjugated π system absorbing and re-emitting with a cooler hue that halos the surrounding amber chains in cold cerulean — and a buried methionine sulfur catches the ambient glow and returns it as a sharp metallic gleam. Far at the edge of perception, perhaps fifteen to twenty ångströms distant, the tight packing loosens almost imperceptibly and a diffuse cool blue-grey haze bleeds inward: the hydrophilic protein surface, where polar side chains surrender to the hydrogen-bonded disorder of bulk water, sealing this amber core from the aqueous world as completely as an insect sealed inside fossil resin.

The surface stretching in every direction belongs to a T=3 icosahedral plant virus capsid just 28 nanometers across — yet from this vantage it reads as a vast crystalline planetoid, its gentle curvature bending toward a molecular horizon tiled in an extraordinary repeating geometry of five-petaled pentameric rosettes and six-petaled hexameric crowns, each petal a discrete coat-protein subunit assembled from precisely folded polypeptide chains. The capsid is built from 180 identical coat-protein copies whose quasi-equivalent contacts lock the icosahedral lattice together through salt bridges flickering red and blue at subunit seams — aspartate oxygen clouds in vivid crimson, lysine nitrogen halos in cool cobalt — while hydrophobic inter-subunit zones radiate a smoldering amber warmth from nonpolar residues buried against the entropic pressure of surrounding water. Concave receptor-binding depressions pool in deep cobalt and indigo between the rosettes, their geometry geometrically tuned to complementary molecular partners drifting through the surrounding cytoplasmic medium, while immunodominant loop summits crest in luminous yellow-white where the protein surface is most exposed and antigenically active. Across every protrusion and ridge, a structured hydration layer two to three nanometers thick coats the outer surface in a quasi-crystalline veil of hydrogen-bonded water molecules, their oxygen atoms scattering the ambient electronic glow into opalescent blue-white highlights — not ornament, but a thermodynamically integral shell that shapes how this protein world is recognized, bound, and ultimately invaded.

Looking directly down the axis of the collagen triple helix, the viewer is drawn into a spiraling corridor of three intertwined polypeptide chains — amber, teal, and jade — coiling away into a warm molecular haze with the hypnotic precision of a braided rope frozen at the boundary between chemistry and architecture. This is collagen in its fundamental form: three left-handed polyproline-II helices wound together into a right-handed supercoil, held in register by a network of interchain hydrogen bonds that shimmer between the strands like gold filaments under tension, supplemented by aquamarine hydroxyproline-water bridges that cool the palette and stitch additional order into the assembly. The extraordinarily tight central column — where glycine residues from all three chains converge with Cα–Cα spacings near 3.9 ångströms — is only possible because glycine carries no side chain, its single hydrogen atom the only geometry small enough to fit; any substitution would rupture the helix entirely. Projecting outward from each strand, the ribbed pyrrolidine rings of proline and hydroxyproline create a densely corrugated exterior surface whose repeating geometry reflects the precise 8.7 ångström helical pitch, reasserting a three-fold rotational symmetry every fraction of a nanometer down the receding tunnel. At this scale, thermal motion is not background noise but a physical presence — the soft focus deepening toward the far end of the helix is not optical aberration but the true consequence of femtosecond bond vibrations, the molecular architecture perpetually alive at the edge of its own stability.

Looking straight down the axis of an amyloid-β fibril, the viewer enters an architectural corridor of almost hallucinatory regularity — parallel β-sheet layers stacked with sub-nanometer precision recede into apparent infinity, each plane of peptide backbone an exact structural echo of the one before it, separated by intervals of roughly 4.7 ångströms governed entirely by hydrogen-bond geometry rather than any external template. Those hydrogen bonds themselves appear as luminous amber-gold rungs spanning laterally between adjacent strands, their directionality perpendicular to the recession axis, tracing a ladder whose terminus is never reached — each N–H donor locked to a carbonyl oxygen acceptor with a bond distance near 2 Å, the quantum mechanical overlap of molecular orbitals rendered as warm, continuous luminescence. At the absolute center of the abyss lies the steric-zipper core, a seam of profound darkness where nonpolar side chains — leucines, isoleucines, valines — interdigitate across the protofilament interface with van der Waals contact so complete that bulk water is entirely excluded, the two apposed β-sheet faces held together by London dispersion forces across a boundary that is, at the molecular level, a seamless coalescence of complementary hydrophobic surfaces. Flanking this dry obsidian corridor, the outer fibril surface shimmers with a blue-white corona of ordered solvation water, each molecule oriented by the polar backbone beneath, hydrogen-bond lifetimes of one to ten picoseconds creating a nacreous thermal shimmer at the liquid-ordered interface. The overwhelming sensation is one of self-templating inevitability: this structure assembled itself one molecular layer at a time, each incoming peptide recognizing and replicating the cross-β geometry of its predecessor with a fidelity that feels less like biology than like crystallographic law made animate.
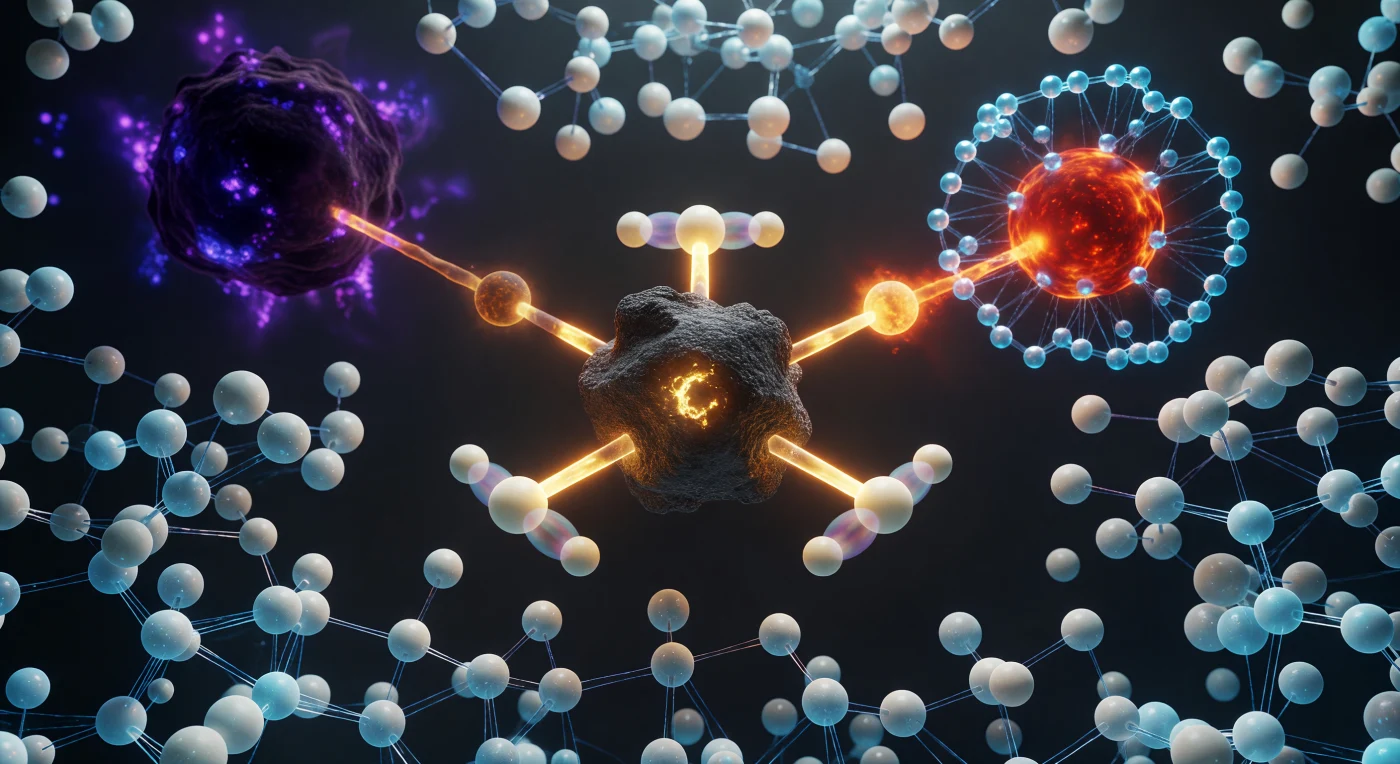
You hang suspended at the apex of a reaction that has not yet decided its own outcome — a moment so fleeting it exists only as a mathematical saddle point on the potential energy surface, lasting on the order of a single bond vibration, roughly ten femtoseconds. The central carbon atom, caught in trigonal bipyramidal transition-state geometry forbidden by equilibrium chemistry, holds three hydrogens locked in a perfect 120° equatorial plane while a nucleophilic oxygen closes from one pole at 2.0 Å and a bromine leaving group stretches away at 2.3 Å, both partial bonds at half-order, meaning neither fully formed nor broken — a condition quantum mechanics permits but classical intuition refuses. Around the carbon's equatorial hydrogens, ghostly translucent twins hover in near-coincident superposition, the signature of quantum tunneling probability distributions that allow hydrogen nuclei to sample classically forbidden positions even at this frozen instant. The surrounding solvent is not empty space but a pressing crowd of water molecules reorganizing their hydrogen-bond network in real time, their cyan halos of electron density straining and reforming to stabilize the emerging charge separation radiating outward from the reaction axis — because at this scale, solvation is not a background condition but an active participant in determining whether the reaction proceeds at all.

You are floating inside a crystalline labyrinth so narrow that the tunnel walls press in from every side at the distance of a single molecular diameter, the entire corridor — an ellipse barely 5.3 by 5.6 ångströms across — built from corner-sharing silicon-oxygen tetrahedra whose pale grey silicon nodes and vivid red oxygen bridges repeat with the unrelenting precision of a mineral crystal grown atom by atom over geological time. Ahead, successive ten-membered-ring portals recede in perfect perspective like the nave of a cathedral whose arches are made of covalent bonds rather than stone, and at a crossroads thirty ångströms forward, a second sinusoidal channel arrives at ninety degrees, creating a molecular junction where diffusing guests must squeeze through a bottleneck barely wider than a water molecule; scattered across these walls, intensely bright Brønsted acid sites mark the aluminum-substituted nodes where a single hydroxyl proton — a welding-arc pinpoint of reactivity — waits to protonate any hydrocarbon that approaches. ZSM-5 zeolite is a microporous aluminosilicate whose precisely engineered channel topology makes it one of the most commercially important catalysts on Earth, used industrially to crack petroleum and produce gasoline, its pore geometry exerting shape-selective control over which molecules can enter, react, and exit — a molecular sieve whose discrimination operates at the ångström level. Golden, space-filling hydrocarbon molecules pack the channel at near van der Waals contact with the oxygen-lined walls, their amber volumes conforming to the sinusoidal corrugation with an almost mechanical fit, held in place not by chemical bonds but by the cumulative whisper of London dispersion forces summed across an entire crystalline surface.
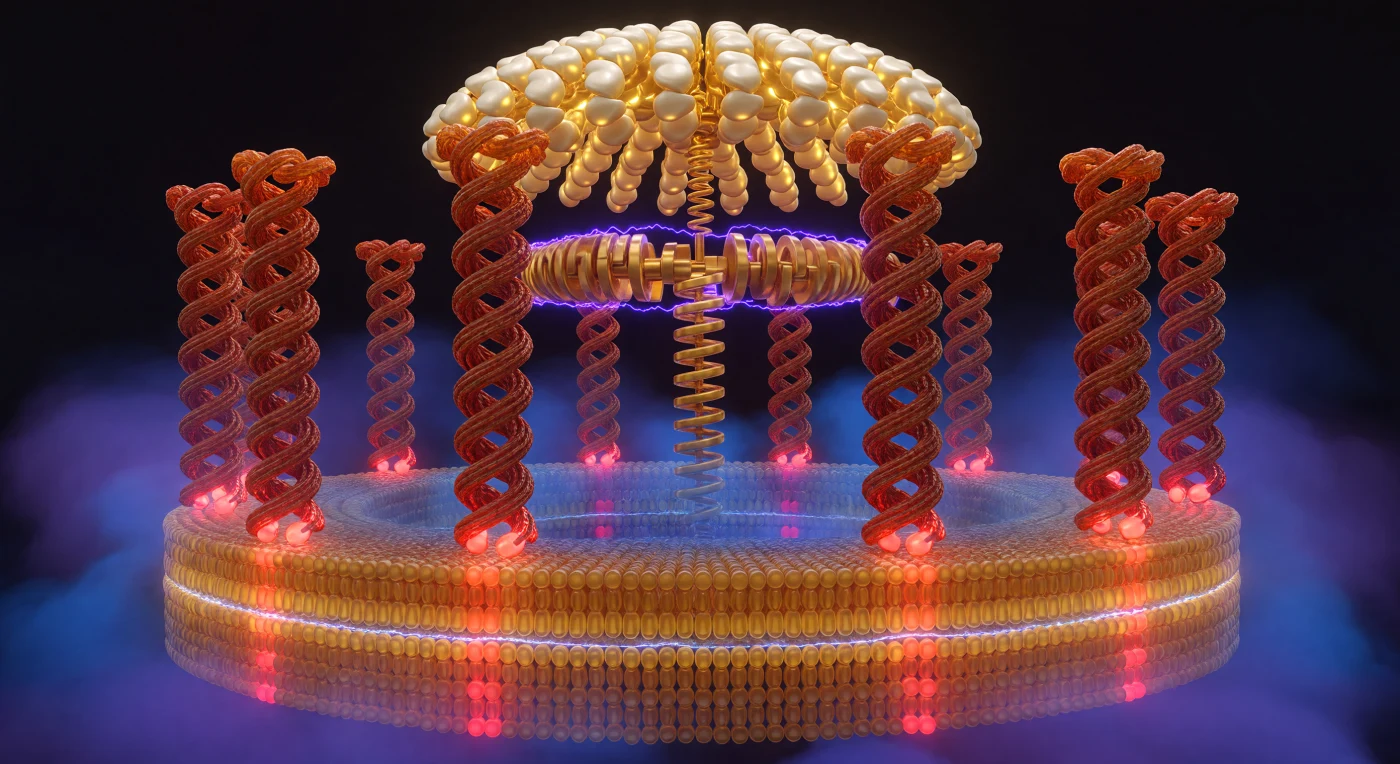
You are standing inside the rotating core of one of the most ancient molecular machines on Earth, enclosed by ten massive α-helical columns of the ATP synthase c-ring that curve away from you on all sides like the interior of a living, breathing colosseum — each column close enough to touch with both hands simultaneously, their terracotta and burnt sienna protein surfaces carrying the subtle spiral ridging of peptide backbone geometry. At the base of each column, individual glutamate residues toggle between blazing crimson deprotonation and muted brick neutrality, marking the precise sites where protons from the intermembrane space bind and release to drive rotation — a cycle powered by the proton-motive force, a 150–200 millivolt electrochemical gradient that saturates the surrounding space as a compression of cobalt and violet light on one face of the membrane and a releasing indigo calm on the other. The lipid bilayer engulfs you at the waist, its amber outer leaflet and blue-tinted inner leaflet meeting at a hard hydrophobic seam that the rotor columns pierce like temple pillars emerging from floodwater, the entire assembly embedded in a membrane only five nanometers thick. Overhead, the F₁ catalytic dome arches in pale gold and moonstone ivory, its asymmetric gamma-subunit camshaft descending into your axis with mechanical inevitability — the eccentric crankshaft that translates the torque of proton-driven rotation into the phosphate bond energy of ATP, a coupling so efficient that it synthesizes roughly three ATP molecules per full revolution, a molecular engine that has been turning without pause for four billion years.
