Scientific confidence: High

You are suspended inside a sealed geological darkness, compressed into the hydrophobic core of a folded globular protein somewhere between two and five nanometers from any solvent boundary, surrounded on all sides by the interlocking bulk of leucine, valine, and phenylalanine side chains packed at roughly 75% density — a figure that rivals the packing efficiency of crystalline minerals, yet achieved entirely through thermodynamic self-organization. The immediate surfaces press close as softly luminous grey-ivory spheroids, their electron-cloud boundaries touching without merging, held apart by quantum mechanical repulsion operating in fractions of a kilojoule, while flat obsidian discs of phenylalanine rings slice the confined space into sharp-edged alcoves whose shadows are absolute, cast not by light but by the total absence of electron density beyond each aromatic plane. Scattered through this achromatic matrix, two or three methionine sulfur atoms return the ambient contact-glow as deep sulfur-yellow embers — the only warm color in the core, marking atoms whose polarizability makes them slightly more luminescent within the van der Waals field than the surrounding carbon and hydrogen. Far beyond the immediate press of side chains, perhaps ten atomic diameters toward the protein's outer surface, the packing loosens imperceptibly and a cold aqueous blue-green light seeps inward through gaps between secondary structural elements — the faint electromagnetic signature of bulk solvent and churning water dipoles, reaching this buried interior like bioluminescence filtered through still ocean depth, confirming that this core is not a void but a sealed, thermodynamically stable interior whose tectonic quiet hums with sub-angstrom thermal vibrations at 310 Kelvin.

You are looking inward along the ribosomal peptide exit tunnel, a corridor roughly 100 ångströms long and barely 15 ångströms across at its narrowest constriction, its walls built from interlocking rRNA helices whose sugar-phosphate backbones curve and ridge like polished horn under the cold blue-white glow bleeding in from the peptidyl transferase center ten nanometers ahead. Every surface here is probabilistic rather than hard — the boundary between rRNA wall and the dense fog of thermally agitated water molecules is a negotiated gradient of electron density, and magnesium and potassium counterions sit in the major-groove indentations like bright silver-white specular points, each one haloed by a shell of rigidly oriented water dipoles that extends its electrostatic influence another ångström or two into the solvent. Threading toward you from that catalytic hearth is the nascent polypeptide itself, its backbone alternating lime-green and warm amber along each peptide bond, its hydrophobic side chains making fleeting van der Waals contact with the ridged nucleotide platforms lining the tunnel walls before thermal motion — occurring on picosecond to nanosecond timescales — peels them away again. The entire passage vibrates at the threshold of perception, every wall, chain, and water molecule executing its own quantum of Brownian motion, the tunnel functioning less as a static architectural form than as a living, electrostatically sculpted channel through which the first folding decisions of a new protein are continuously being made in amber shadow and catalytic blue fire.

You are hurtling through a corridor so narrow it barely spans twenty atomic diameters, the walls alive with the restless blur of electron density rather than any solid surface — a vertiginous flight down the major groove of B-form DNA, where the 2.2-nanometer-wide channel spirals gently rightward in a clockwise helix, its geometry dictated entirely by the Watson-Crick base-pairing rules that enforce precise geometric complementarity between every amber adenine and sage-green thymine, every deep-teal guanine and sky-blue cytosine stacked 0.34 nanometers apart in aromatic platforms whose overlapping π-electron clouds shimmer with translucent violet-indigo interference. The phosphate-sugar backbone rises on both sides like a braided railing of orange-tan sea glass backlit from within, its negative charges drawing clusters of magnesium cations as hard white-blue sparks ringed by shells of oriented water, and sodium ions as soft gold halos drifting through the Debye screening layer — that milky, electrostatically charged fog where polarized water dipoles ripple in slow aurora-like wavefronts of blue-white light that wash rhythmically through the groove. Thermal bombardment is total and inescapable: 0.28-nanometer water molecules slam the base-pair floor in a continuous low tremor, each collision a flicker of white light instantly swallowed by the aqueous glow, reminding you that at this scale there is no stillness, only the ceaseless molecular violence of room-temperature solvent pressing in from every direction at once.

The view opens onto two vast opposing surfaces separated by a narrowing void barely wide enough for a few water molecules — the closing interface between an antibody's Fab fragment and its target antigen, resolved here into overwhelming sculptural detail. From behind and above, the beta-sandwich body of the Fab looms like a pewter massif, its laminar ridges of tightly hydrogen-bonded beta-strands giving way at the crown to six CDR loops surging forward like enormous architectural fingers: the amber H3 loop arcing in a long confident curve, the teal L3 reaching in from below, the four flanking loops framing them in graduated silvers and dusty mauves to form a cupped amphitheatre of complementary molecular geometry. Across the narrowing gap, the antigen epitope curves away like a convex planetary horizon, its ochre and terracotta surface contoured with uncanny precision to receive the approaching loops — the product of evolutionary or affinity-matured shape complementarity operating across a contact zone of roughly five by six nanometres. Water molecules are being expelled from this drying interface in real time, radiating outward as brief opalescent sparks of dipole-reorienting thermal energy, while cyan hydrogen-bond bridges flicker and consolidate across the remaining Ångström gap — luminous blue-white threads of shared electron density stabilizing one by one as electrostatic, van der Waals, and hydrophobic contributions sum toward the final binding free energy. The surrounding medium presses in from every direction as warm sapphire solvent haze, a thermally turbulent ocean of molecular bombardment that makes the slow, deliberate geometry of this molecular recognition feel all the more improbable and precise.
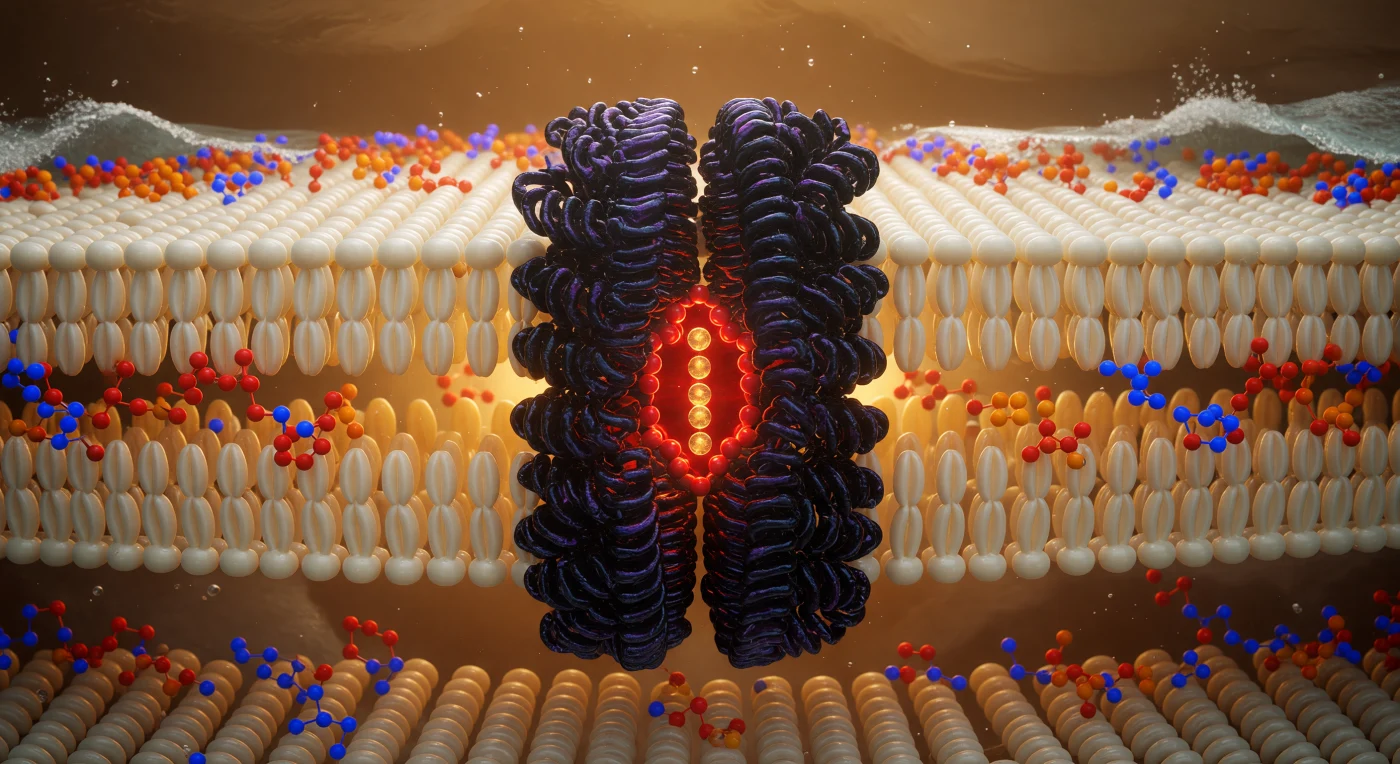
You are suspended at the geometric heart of a living membrane, hovering in the hydrocarbon midplane where phospholipid tails from opposing leaflets interdigitate in a dense, shimmering forest of ivory-gold carbon chains — each one a softly luminous cylinder roughly 0.4 nanometers across, packed so tightly that the interstitial spaces between them read as barely-there shadows in the ambient electrostatic glow. This hydrophobic core, spanning roughly 3–4 nanometers in total thickness, is the thermodynamic engine of membrane integrity: water is excluded not by any hard barrier but by the collective entropic cost of disrupting the hydrogen-bond network outside, making this oily interior a sanctuary of van der Waals stillness punctuated only by the ceaseless thermal writhing of unsaturated chain kinks. Rising through that fog directly ahead, the potassium channel — a tetrameric integral membrane protein whose transmembrane helices are stitched into the bilayer by hydrophobic matching, their nonpolar residues interdigitating seamlessly with the surrounding lipid tails — presents itself as a cathedral column of deep purple-black density, its fourfold symmetry resolving at the pore axis into the iconic selectivity filter: a precise angstrom-scale cage of backbone carbonyl oxygens that strips hydration shells from K⁺ ions and passes them in single file at rates approaching 10⁸ ions per second, burning red-orange with concentrated electron density. Above and below, two luminous headgroup shorelines mark the polar-nonpolar interfaces — phosphorus nodes flaring cadmium-orange, choline nitrogens pulsing cobalt-blue — while beyond them, invisible from here, the aqueous world hammers the charged surface with water molecules at hundreds of meters per second, the turbulent exterior pressing against this amber-lit interior like an ocean held at bay by chemistry alone.

You drift just above a vast corrugated plain whose amber-parchment ridges sweep to every horizon in alternating directions — the backbone strands of an antiparallel beta-sheet, each one a dense ribbon of covalent bonds carrying the faint translucent sheen of electron-density clouds rather than any hard surface. Cherry-red carbonyl oxygens crown each ridge crest like garnets set into old bone, while pale amide nitrogen tips line the valley floors, and between every pair of adjacent strands a repeating series of cyan hydrogen bonds arches laterally — ghostly filaments of electrostatic probability glowing at 2.9-ångström intimacy, collectively lacing the plain into molecular chainmail of extraordinary regularity. The light here is directionless, a diffuse cold-white glow sourced from the surrounding aqueous medium rather than any sun, catching every backbone ridge top and leaving the valleys in warm amber shadow, while at floor level van der Waals contacts pack the molecular surface so tightly that no vacuum remains, the texture almost marbled. At the sheet's far edge, the disciplined corrugations dissolve abruptly into writhing red-orange connector loops — disordered turn regions frozen mid-fluctuation, their surfaces knobbed with side-chain protrusions — before the entire structure blurs into the blue-gray ionic haze of bulk solvent, a horizon that breathes with the constant reorientation of water dipoles too fast to resolve.

You find yourself suspended at the geometric center of a sealed molecular vault — a spherical enclosure roughly eight nanometers across whose pale, smoothly corrugated walls curve away from you in every direction with the silent completeness of the inside of a pearl, each segment of the GroEL barrel subtly articulated, its hydrophilic inner lining coated in outward-bristling side chains that catch a sourceless cool blue-white luminescence as though the protein itself radiates aqueous light. Overhead, seven rounded subunits of the GroES co-chaperonin lock together into a shallow dome, their fitted interfaces hairline seams of darker pewter, sealing this chamber against the cytoplasmic chaos outside — a molecular airlock designed by four billion years of selection pressure to give unfolded proteins a second chance. At the chamber's center, three subjective meters away, a misfolded substrate protein drifts in slow Brownian suspension, a crumpled mass of beige-amber polypeptide whose disordered loops fray probabilistically into the surrounding aqueous haze, glowing with a faint organic warmth entirely unlike the antiseptic radiance of the walls enclosing it. This is the GroEL-GroES chaperonin system in action: by sequestering the substrate within a chemically buffered, hydrophilic microenvironment and coupling ATP hydrolysis to cyclical enclosure and release, the complex suppresses off-pathway aggregation and grants the chain the isolation and time — milliseconds to seconds — needed to navigate its folding energy landscape toward a stable, functional conformation.

You are suspended just a few nanometers above the replication fork, close enough that the aqueous medium surrounding you feels less like open space and more like a warm, faintly luminous gel — water molecules hammering you from every direction at hundreds of meters per second, their flickering blue-white dipoles vanishing before they fully resolve. Below you, the CMG helicase hexamer dominates the scene like a quartzite millstone fifteen nanometers across, its interlocked protein subunits roughened with alpha-helical ridges and beta-sheet plateaus, its inner channel radiating an intimate ochre phosphorescence as it actively pries apart the incoming parental double helix — that deep-navy, architecturally columnar B-form DNA feeding in from the right, its pi-stacked aromatic core dark as polished jet, its major groove wide enough to feel like a carved stone nave. From the unwinding junction, two single-stranded template threads unspool into the forward space: the leading-strand template in saturated teal, phosphorescent along its backbone, and the lagging-strand template in molten gold-amber, its exposed bases swaying visibly in the Brownian current as they lose their hydrogen-bond partners in soft probabilistic bursts of pale warmth. Ahead on the leading strand, the DNA polymerase clamps the nascent duplex in a grey-ochre articulated grip, firing sharp orange-white pyrophosphate blooms with each nucleotide incorporation — femtosecond flares that dissipate instantly into the thermal haze — while magnesium ions streak through invisible electrostatic channels as silver-white sparks, and single-stranded binding proteins drift in the middle distance like pale translucent jellyfish, their disordered loops trailing in the current of a machine replicating the genome at roughly a thousand base pairs every second.

You are looking up through a forest of protein columns that rise around you in rhythmic, clockwise spirals, each pillar a right-handed alpha-helix just 1.2 nanometers across and six nanometers tall, their surfaces alternating between warm amber ridges and deep gold grooves where every backbone carbonyl and peptide bond leaves its own tiny bright or shadowed node in the molecular grain. These are coiled-coil assemblies — paired helices drawn into intimate contact along a repeating seam of hydrophobic leucine residues that project inward like interlocking bronze spokes, held together not by covalent bonds but by the cumulative whisper of van der Waals attraction, while outward-facing lysine and arginine side chains flare into the surrounding solvent in saturated crimson and electric blue, their charged tips bleeding diffuse electrostatic halos into the aqueous medium. The space between columns is cathedral-narrow, barely wide enough for a few water molecules to thread through, and along the hydrophobic seam water is excluded entirely, creating a zone of dense, oily shadow that feels gravitationally heavier than the bright solvent corridors to either side. Above, the aqueous sky is a luminous cerulean aerogel, its 0.28-nanometer water molecules colliding and reorienting so rapidly that their hydrogen-bond networks register only as a constant gossamer shimmer, flickering in and out across picosecond intervals. The forest recedes through twenty nanometers of molecular haze, each successive rank of helical columns softening into cooler blue-grey as the medium scatters whatever ambient illumination permeates this world, the leucine-contact seams tracing elegant herringbone diagonals that dissolve quietly at the limit of molecular visibility.

You are suspended at the dead center of the fibril axis, looking down a tunnel of four protofilament lobes that sweep outward from you like the petals of a gothic flower, each wing a curved wall of beta-strands stacked in relentless 4.7-ångström increments — a spacing so precise it functions less as a measurement than as the structural heartbeat of an entire category of molecular pathology. These are amyloid fibrils: cross-beta assemblies formed when once-soluble proteins collapse out of their native conformations and lock into hydrogen-bonded sheets that propagate laterally with a thermodynamic permanence rivaling crystalline mineral. At the core, between the four protofilaments, a steric zipper seals the structure from within — interdigitated side chains meshed in near-contact, van der Waals surfaces touching across a gap so narrow that bulk water is excluded entirely, replaced by a dry, amber-glowing compression of electron density that drives stability through hydrophobic burial and geometric complementarity too exact to be accidental. At the outer frontier, glutamate and lysine residues reach into the surrounding solvent haze — electrostatically charged sentinels creating the dielectric fringe that both stabilizes the assembly against lateral aggregation and presents the surface chemistry that living systems cannot easily degrade. The entire structure, roughly 10 nanometers across, extends for hundreds of nanometers along the axis receding above you: at this scale, that distance reads as geological, a molecular colonnade built from the slow catastrophe of misfolding.
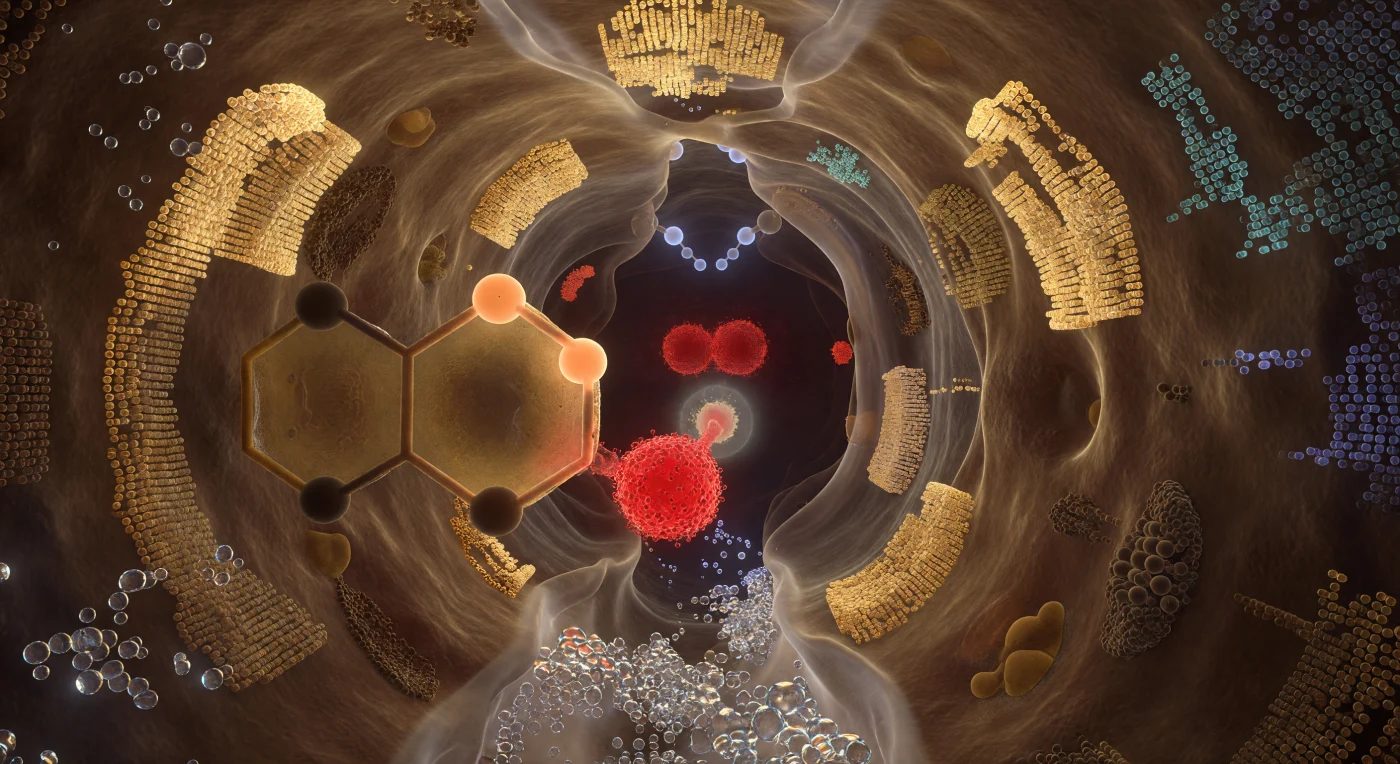
You are standing inside the reaction chamber itself — a cleft no wider than a few atomic diameters, its amber-shadowed walls built from the compacted van der Waals surfaces of hundreds of tightly packed side chains, their outermost electron shells dissolving into quantum probability fog at every contact boundary. The catalytic triad dominates the space like three geological monuments: the Ser195 oxygen burns as a crimson node just angstroms from the substrate's scissile bond, its lone-pair density straining forward under electrostatic compulsion; the imidazole ring of His57 fills your lateral field as a vast aromatic panel of amber-stained glass, a shuttled proton frozen mid-transfer between its two nitrogen atoms in warm apricot suspension; and behind both, Asp102 anchors the entire relay as a deep garnet mass, its resonance-equivalent oxygens pulsing with delocalized negative charge that stabilizes the proton shuttle like bedrock. Above the developing tetrahedral intermediate, the oxyanion hole opens as a vaulted recess, two backbone N–H donors projecting pale-blue hydrogen atoms inward like pincers, their partial positive charges casting cold electrostatic light onto the carbonyl carbon below in a geometry evolution has refined across hundreds of millions of years of serine protease history. The entire pocket breathes not in mechanical rhythm but in probabilistic flux — every surface simultaneously solid and spectral, every bond length a statistical mean, the chemistry suspended at the precise Boltzmann-weighted instant before the covalent intermediate collapses and a peptide bond ceases to exist.

You float three nanometers from a structure that fills your entire field of view from edge to edge — a triple-stranded polypeptide cable barely 1.5 nanometers wide, its three chains wound in a slow right-handed superhelix of warm ivory, pale gold, and sun-bleached tan, each strand inseparable from the others yet distinct, braided with the patience of rope twisted over geological time. At every third residue along each chain, the rigid pyrrolidine rings of proline residues jut like pale grey knuckles into the helical grooves, their flat cyclic geometry a structural necessity: collagen cannot tolerate bulkier side chains at these positions, and it is precisely this enforced rigidity that gives the triple helix its remarkable tensile strength — capable of withstanding loads that would rupture most synthetic polymers of comparable cross-section. Amber hydroxyproline hydroxyl groups project outward at irregular intervals, each one cradling a single water molecule in a hydrogen bond, and together these contacts nucleate a ghostly first hydration shell of pale crystalline blue that encases the entire cable in near-perfect tetrahedral order at 2.8-ångström spacing, a frozen breath of solvent locked by dipole geometry against the charged backbone. Ahead of you the cable recedes for what feels like an infinite corridor — hundreds of nanometers dissolving into luminous aquamarine molecular fog, the collective electron haze of ten thousand water molecules per cubic nanometer scattering thermal radiation into directionless cool light — the full length of a single tropocollagen molecule extending toward an horizon you cannot reach.
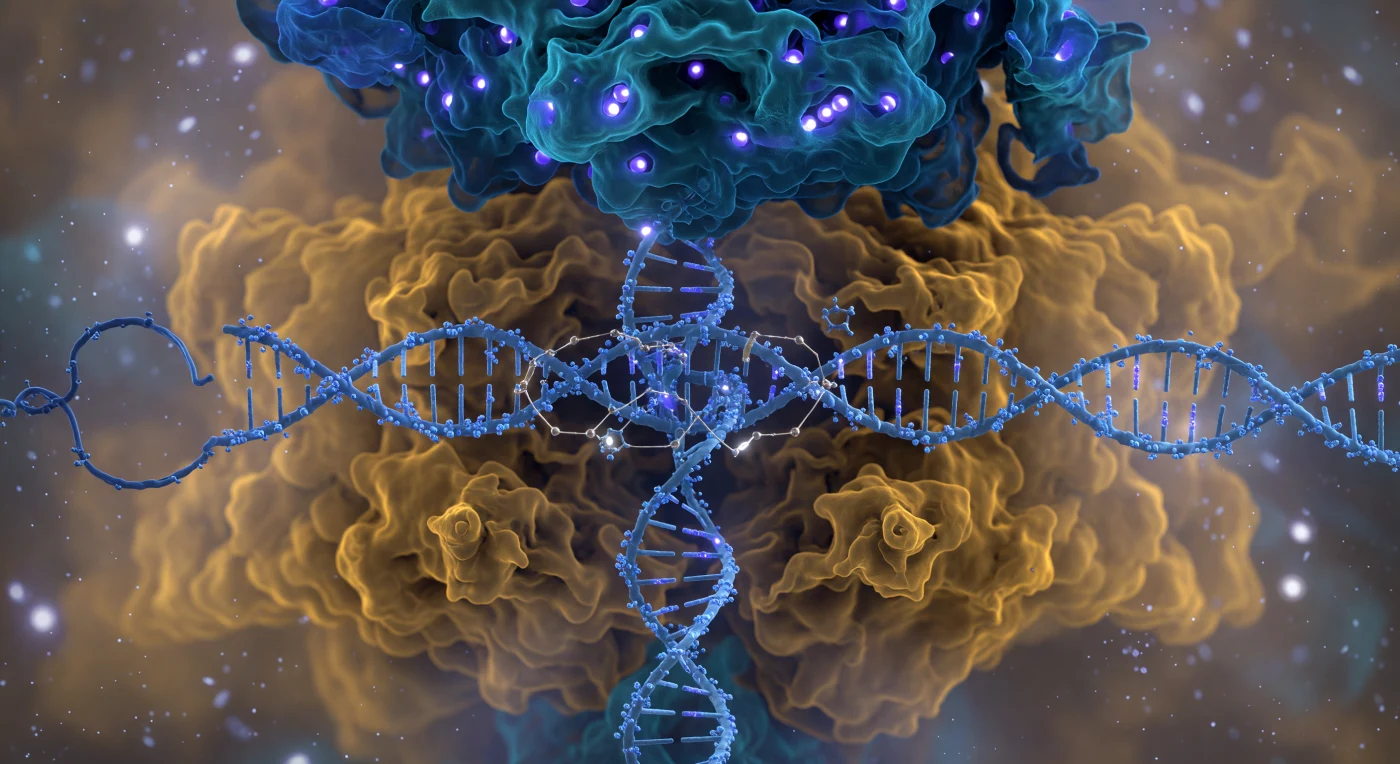
You are suspended five nanometers from the face of one of the most consequential molecular machines in biology, and the entire visual field is consumed by the Cas9 complex — a bilobed protein nearly ten nanometers across whose cobalt-blue recognition lobe overhangs you like a vaulted cathedral ceiling while the warm amber nuclease lobe swells outward below, both surfaces resolving not into hard edges but into probabilistic electron-density fog, the boundary between molecule and solvent perpetually negotiated rather than fixed. Threading between the two lobes, the ice-blue DNA double helix cuts across your view like a pillar of twisted rope under electrostatic tension, its phosphate backbone nodes glowing with cool violet charge while the displaced non-template strand loops away into the molecular haze — the major groove facing you directly, a shadowed canyon whose base chemistry is being read with extraordinary precision as the electric-cyan guide RNA spacer reaches inward to form R-loop base-pair bridges, each hydrogen bond rendered as a gossamer thread of gold-white light snapping two surfaces into complementary contact. On the rightward face, arginine fingers from the PAM-interacting domain press into the minor groove of the NGG trinucleotide like a key reading a lock, locally unwinding the helix geometry in a distortion you perceive as a visible narrowing of the groove. Deep in the catalytic cleft, paired magnesium ions occupy the HNH and RuvC active sites as brilliant white-hot pinpoints of divalent charge, each caged in octahedral coordination geometry — poised, once full R-loop formation is confirmed, to execute the precisely sequenced phosphodiester cleavages that will sever both strands of the target DNA with an accuracy that has reshaped the life sciences.

You are suspended at the geometric heart of a molecule that refuses to hold a single shape — the conformational ensemble spreading around you like a living fog, a blue-white probability cloud roughly eight nanometers across built from dozens of polypeptide conformers overlaid at near-total translucency, their collective presence suggesting structure without ever committing to it. This is an intrinsically disordered protein, a chain that has abandoned the stable folded architecture of classical structural biology in favor of a dynamic ensemble of configurations, each thermodynamically accessible, none dominant, the whole population sampling sequence space continuously on nanosecond timescales driven by thermal collisions with the surrounding solvent. To your left, a faint amber ribbon momentarily resolves — an alpha-helix flickering into hydrogen-bonded coherence for perhaps a nanosecond, its carbonyl oxygens catching warm internal light before the chain's entropic freedom dissolves it back into the blue-white blur — while nearby a hydrophobic cluster of aromatic residues pulses with brief yellow-gold luminescence, a transient energetic minimum forming and collapsing within microseconds as phenylalanine and tryptophan side chains press into fleeting van der Waals contact. The water is not background here but the dominant force of existence: spheroidal molecules barely 0.28 nanometers across hammer every exposed backbone segment in relentless thermal agitation, their collective jostling the engine that drives conformational sampling and the diffuse aqueous haze that swallows the ensemble's outermost tendrils beyond fifteen nanometers into statistical invisibility.

Looking up from below the stem base, you are confronted by a spiraling tower of interlocked amber and copper rings that fills the entire sky above — the A-form double helix of an RNA hairpin, its geometry noticeably more compact and inclined than DNA, its 2.3-nanometer width tapering as it climbs through layered aqueous haze toward the baroque flowering crown of the GNRA tetraloop far overhead. The ribose-phosphate backbone rises in two intertwined strands of burnished bronze-ochre, each ribose unit bristling with a small copper-bright 2'-hydroxyl antenna — those outward-pointing oxygen groups that chemically distinguish RNA from DNA, vibrating softly within their local energy wells and catching the ambient light with a warmer glow than the surrounding solvent. Crowding in from every direction, water molecules manifest as semi-translucent opalescent spheroids in perpetual agitated motion, their oxygen atoms flickering with cold blue sparks, while sodium ions streak past as hard silver-white points trailing brief hydration wakes and magnesium ions hover in warm gold-green clusters close to the phosphate groups, electrostatically tethered to the backbone's dense negative charge. The base pairs stacked within the minor groove above are dark iridescent sheets of guanine and cytosine whose pi-stacked aromatic electrons delocalize across neighboring rings in shimmering plum and teal, and the whole structure vibrates invisibly at femtosecond rhythms even as this instant holds it in monumental, luminous stillness.
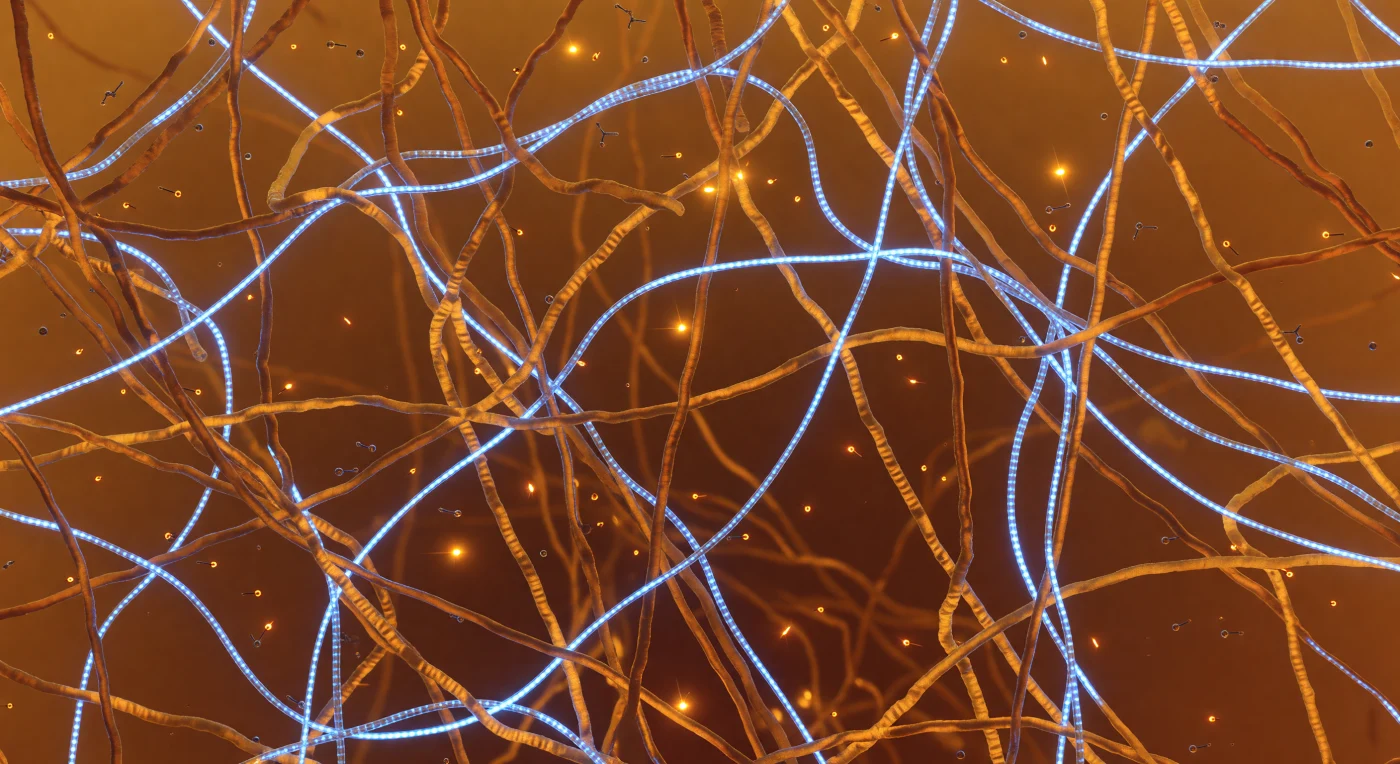
You are standing inside the condensate itself, surrounded on every side by a dense amber lattice of disordered low-complexity domain protein chains — intrinsically unstructured polypeptides that have demixed from the surrounding cytoplasm through liquid-liquid phase separation, concentrating here into a viscoelastic gel-like droplet stabilized not by any membrane but by the collective weight of transient molecular contacts. The cation-π interactions you witness as fleeting amber sparks — tyrosine aromatic rings pressing momentarily against arginine guanidinium groups — are among the primary driving forces that hold this condensate together, each bond lasting only nanoseconds before thermal energy dissolves it and another forms nearby, so that the network is simultaneously permanent at the mesoscale and completely fluid at the molecular scale. Threading through the amber polymer mesh, the RNA strands glow with the structured luminescence of their phosphate backbones and stacked aromatic bases, their 1.5-nanometer diameter making them substantial cables within a mesh that opens only 5 to 15 nanometers between protein strands — pores just wide enough for small metabolites like ATP to diffuse through in pure Brownian motion, without any directed transport, simply carried by the thermal agitation of surrounding water molecules striking them at hundreds of meters per second. The fog that swallows your depth of field within five nanometers is not optical illusion but physical reality: this condensate is so molecularly dense that there is no meaningful "open space," only gradations of polymer concentration and the warm, diffuse luminescence of a living chemical environment where the boundary between structure and solution has deliberately, functionally dissolved.
